How to run MAGeCK Count on Latch
- Find MAGeCK Count in your Workspace
- Find MAGeCK Count in “All Workflows” and open the workflow
- Enter the parameters for MAGeCK Count
- First add your Sample Labels, the labels you add should correspond to the Sample FastQ files you will give MAGeCK. Ex. “L1”, “CTRL”
- Then add your Sample FastQ file, these should correspond order wise to the Sample Labels you gave in the previous step.
- If you have technical replicates for a sample, add them within the same box.
- Then select your List Sequence File.
- You can learn more about the format of this file below.
- Then fill out the Output Prefix and Output Location and click Launch Workflow.
- Within no time your results will show up in the Data tab!
FYI
- If you want to run multiple executions of this workflow click the large plus button at the bottom of the parameters to add an additional execution of the count workflow.
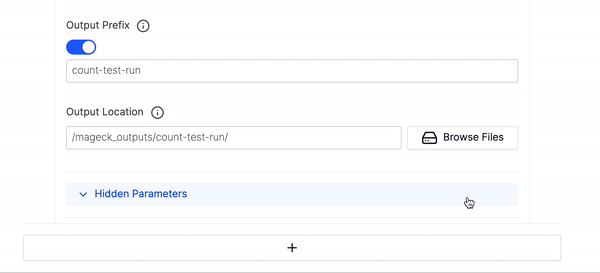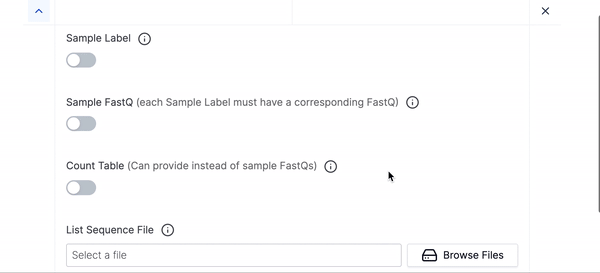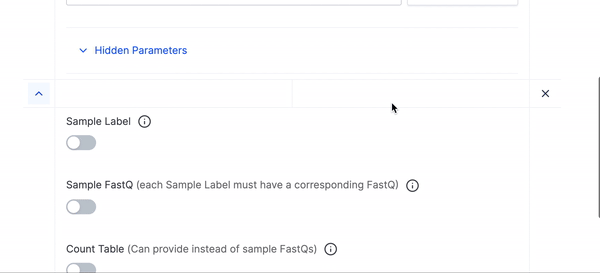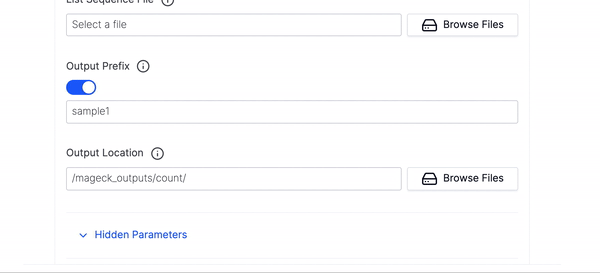
- We have hidden many of the optional parameters under Hidden Parameters, you can click that if you would like to fine tune your execution run or want to use any of the advanced parameters.
Required Parameters
Sample Labels
- The labels of each sample, these labels will be used to specify whether the samples are treatment or control in later MAGeCK steps
- This defaults to sample1, sample2, etc… but would recommend specifying them yourself in this step as they are needed in later subcommands.
Fastq reads for each sample
- The reads for each sample and should correspond to each sample label, each sample can have Technical Replicates added as well.
- Accepted Files
- fastq
- fastq.gz
- SAM/BAM
- If the sample reads are paired ends, the 2nd Fastq can be added to 2nd FastQ for Paired End Reads in the hidden parameters accordion. The files given here must correspond orderwise to the files given in the first Fastq parameter.
- If you have Biological Replicates treat them as separate samples in MAGeCK Count and then you will be able to specify them as such in the MAGeCK Test and MAGeCK MLE steps when doing analysis.
List Sequence File
- A file containing the list of sgRNA names, their sequences and associated genes. When starting from FASTQ, FASTQ.GZ or BAM files, MAGeCK needs to know the sgRNA sequences and targeting genes.
- Accepted Files
- .tsv
- .csv
- .txt with tab or comma separated values
- Example: - You can download (right click to download) an example txt tab separated library file here:
library.txt
87.7KB- There are three columns in the library file: the sgRNA ID, the sequence, and the gene it is targeting.
| sgRNA ID | Sequence | Gene |
|---|---|---|
| s_10007 | TGTTCACAGTATAGTTTGCC | CCNA1 |
| s_10008 | TTCTCCCTAATTGCTTGCTG | CCNA1 |
| s_10027 | ACATGTTGCTTCCCCTTGCA | CCNC |
Output Prefix
- The prefix appended to all of the outputted files
Output Location
- The directory where the files produced by this subcommand will be placed. A path can either be selected or if a new path is typed in field Latch will automatically create the folders in the data viewer.
Hidden Parameters
FastQ Parameters
2nd FastQs for Paired End Reads
- The 2nd fastqs for paired end reads for each sample. These should correspond to each sample label, and each sample can have technical replicates added as well.
Quality Control Parameters
Day Zero Label
- Specifying this will turn on the negative selection quality control and specify the label as the control sample (usually day 0 or plasmid). For every other sample label, the negative selection quality control will compare it with day0 sample, and estimate the degree of negative selections in essential genes.
Length of 5’ End Read Trimming
- Length of trimming the 5’ of the reads. Default 0
Disable Discarding of sgRNAs Containing ‘N’ in FastQ Reads
- Enabling this will count sgRNAs with Ns. By default, sgRNAs containing Ns are discarded by MAGeCK
- It is expected that the first few thousand reads in an Illumina sequence fastq file are of comparatively low quality and frequently contain “N”s. An “N” means that the Illumina software was not able to make a basecall for this base. The reads at the beginning and end of the sequence data files originate from the edges of the flowcells, where imaging is more difficult, thus these reads show below average quality which is why MAGeCK by default discards them.
Reverse Complement the Sequences in Library for Read Mapping
- Enabling this has MAGeCK reverse complement the sequences in the library for read mapping. By default read mapping will be performed with the sequences as they are in the library.
Method for Normalization
- By default MAGeCK will use Median normalization.
- Options:
- None: no normalization
- Median: median normalization, default
- Total: normalization by total read counts
- Control: normalization by control sgRNAs specified by the Control sgRNAs option. The median factor used for normalization will be calculated based on control sgRNAs only, rather than all the sgRNAs
Control sgRNAs
- A list of control sgRNAs for normalization and for generating the null distribution of RRA. Alternatively Control Genes can be specified instead of this parameter. This option tells MAGeCK to use provided negative control sgRNAs to generate the null distribution when calculating the p values. By providing the corresponding sgRNA IDs in this parameter, MAGeCK will have a better estimation of p values.
- When using this option, you will need to provide a plain text file just containing negative control sgRNA IDS (one per each line). For example,
Control Genes
- A list of genes whose sgRNAs are used as control sgRNAs for normalization and for generating the null distribution of RRA. Alternatively Control sgRNA can be specified instead of this parameter. There are several issues that you need to keep in mind:
- You should have enough number of negative control guides (>100 recommended) for accurate p value estimation and normalization.
- It is known that for growth based screens, non-targeting controls may lead to high false positives (e.g., Morgens et al. 2017. Use non-targeting controls carefully.
- By default MAGeCK will generate the null distribution of RRA scores by assuming all of the genes in the library are non-essential. This approach is sometimes over-conservative, and you can improve this if you know some genes are not essential.
Use Custom Pathway File For Quality Control (GMT Format)
- The pathway file used for QC, in GMT format. By default it will use the GMT file provided by MAGeCK (mageckQC.gmt.
- More information about GMT format can be found here and a repository for pathway files can be found here.
Output Settings
sgRNA Length
- The length of the sgRNA. The program will automatically determine the sgRNA length from library file, so this parameter should likely be toggled off. If toggled on and given an sgRNA length, will put umapped reads to a file for viewing.
Keep Intermediate Files
- Keeps intermediate files for this subcommand which are the .r, .rmb,.rnw files which can brought into an R software environment to plot the results of the execution
Run Settings
Test Run Using First 1M Records For Each File
Outputs
count.txt
- A tab-separated count table, each line in the table should will the sgRNA name (1st column), the targeting gene (2nd column) and the read counts in each sample. Each item will be separated by the tab (‘\t’).
- For example in the studies of T. Wang et al. Science 2014, there are 4 CRISPR screening samples, and they are labeled as: HL60.initial, KBM7.initial, HL60.final, KBM7.final. Here are a few example lines of the count file:
| sgRNA | Gene | HL60.initial | KBM7.initial | HL60.final | KBM7.final |
|---|---|---|---|---|---|
| A1CF_m52595977 | A1CF | 213 | 274 | 883 | 175 |
| A1CF_m52596017 | A1CF | 294 | 412 | 1554 | 1891 |
| A1CF_m52596056 | A1CF | 421 | 368 | 566 | 759 |
| A1CF_m52603842 | A1CF | 274 | 243 | 314 | 855 |
| A1CF_m52603847 | A1CF | 0 | 50 | 145 | 266 |
- This count file will be used for the count table parameter in MAGeCK Test and MAGeCK MLE workflows.
count_normalized.txt
- A normalized count file. Please forgive me as I’m not really sure what the significance of this file is, but will update this once I figure it out. Or if you can help explain this to me please contact me at nathan@latch.bio.
countsummary.txt
- This file is generated by count command, and summarizes QC measurements of the fastq (or count table) files. Learn more about it from the MAGeCK Wiki.
countssummary.R
- This file contains code that can be executed within the R software environment to plot the data from the count subcommand and create a PDF from it. This file can be used in a program such as RStudio.
countsummary.Rnw
- This file is called by the counts summary.R file and has the specific code for plotting the results.
Log File
- This file contains all of the logs of the execution. This file is mostly a bunch of techno gobbledygook but you can view it to view any errors the execution might have encountered.