How to run CRISPOR on Latch
- Add CRISPOR to your Workspace
- Find CRISPOR in “All Workflows” and open the workflow
- Enter the parameters for CRISPOR
- Enter a single genomic sequence, < 2300 bp, typically an exon
- NOTE: Text case is preserved (e.g. ATCG & atcg both work)
- You can paste multiple sequences >23bp, separated by N characters.
- Avoid using cDNA sequences as input, CRISPR guides that straddle splice sites are unlikely to work.
- Select your genome
- Select from 704 different genomes! Contact CRISPOR support if yours is missing.
- Find links to pre-calculated exonic guides for each genome here on the UCSC genome browser.
- Select a Protospacer Adjacent Motif (PAM)
- Select from ~40 options
- Support for cas9, cas12, casX, & many more
- See notes on enzymes for more info
- Then select the Output Location and click Launch Workflow.
- Enter a single genomic sequence, < 2300 bp, typically an exon
- Within no time your results will show up in the Data tab!
Parameters
Sequence Name
- Just a semantic name for your sequence data
Sequence
- Enter a single genomic sequence,
<2300base pairs, typically an exon
PAM
- Protospacer Adjacent Motif (PAM)
- For most current applications of the CRISPR-Cas system, Streptococcus pyogenes Cas9 nuclease is used and the corresponding PAM is NGG.
- However, you can choose other enzymes and corresponding PAMs from the dropdown box.
Genome
- Select your genome of interest from the list
Output Location
- The directory where the files produced by this workflow will be placed. A path can either be selected or if a new path is typed in field Latch will automatically create the folders in the data viewer.
Outputs
Output 1: Annotated input sequence
The main output of CRISPOR is a page that shows the annotated input sequence at the top and the list of possible guides in the input sequence at the bottom.guide.csv
4.6KB
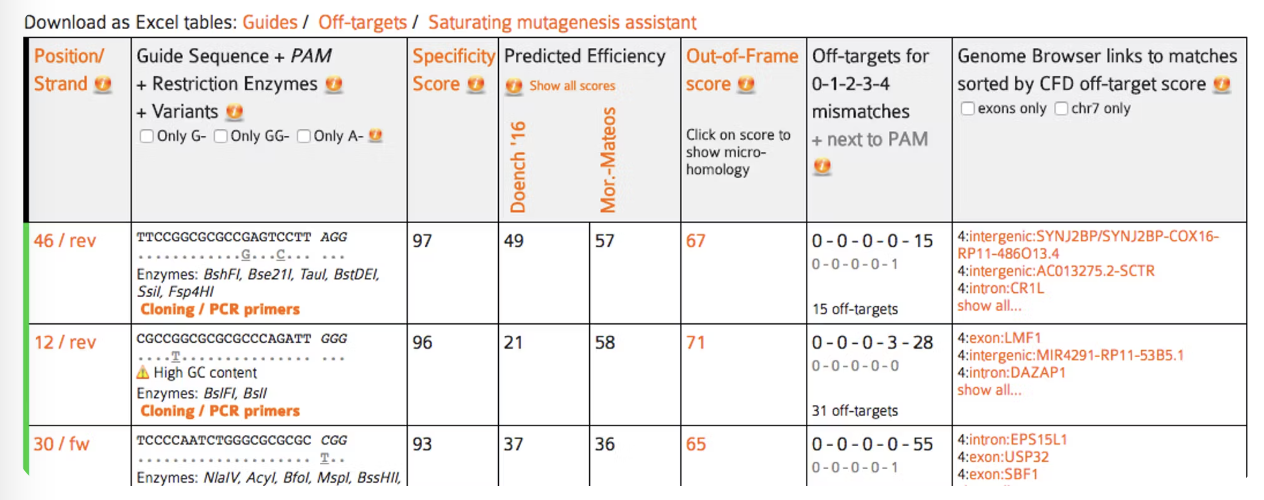
- this is the position of the PAM on the input sequence and the strand, e.g. “13+”
- the sequence of the guide target and the PAM and the link to its “PCR and cloning primers” (see the Primers section
- a prediction of how much an RNA guide sequence for this target may lead to off-target cleavage somewhere else in the genome.
- the efficiency score is a prediction of how well this target may be cut by its RNA guide sequence.
- this score (0-100) is a prediction how likely a guide is to lead to out-of-frame deletions.
- the number of possible off-targets in the genome, for each number of mismatches.
- the locations of all possible off-targets with up to four mismatches, annotated with additional information