Documentation Index
Fetch the complete documentation index at: https://wiki.latch.bio/llms.txt
Use this file to discover all available pages before exploring further.
Our goal with our platform is to bring a one-stop no-code interface to each of these workflows. We would love to hear about how you use this workflow and what you use it for so we can improve the experience along the road.Your feedback means the world to us. At any time feel free to click the chat icon on our platform (bottom left) to leave comments, requests, questions, bugs, or just say hi :)
How to run DECODR Count on Latch
- Add DECODR to your Workspace
- Find DECODER in “All Workflows” and open the workflow
- Enter parameters for DECODR
- Launch the workflow
- Results appear in Latch Data instantly
Parameters
Sample Names
- The names or labels of your samples
Sample Types
- These types are clonal or bulk. The sample type sets a limit on the number of sequences DECODR can find in an experimental trace.
Guide Sequences
- Enter each gRNA (max 2), 17-30 nucleotides. Don’t include the PAM (NGG) in the sequence)
- Example Value: AGTGCTGATCGTACTCAGAGGAC
Donor sequence (optional)
- A sequence of 36 to 300 nucleotides with homology arms at least 18bp long
- Example Value: AGTGCTGATCGTCAGCATGACGTATGACTACTACGTAGTGCTTGCTAGCTGATCG
Wild Type/Control File
- This can be input of one of two ways
- File input: Browse to select your wild type/control file.
- Accepted File Types: .ab1, .fasta, .fastq., .txt
- Text input: Enter the plain text sequence. This value must be at least 300 nucleotides
Experiment Files
- You can upload multiple files here to do a bulk analysis
- Accepted File Types: .ab1, .fasta, .fastq., .txt
Outputs
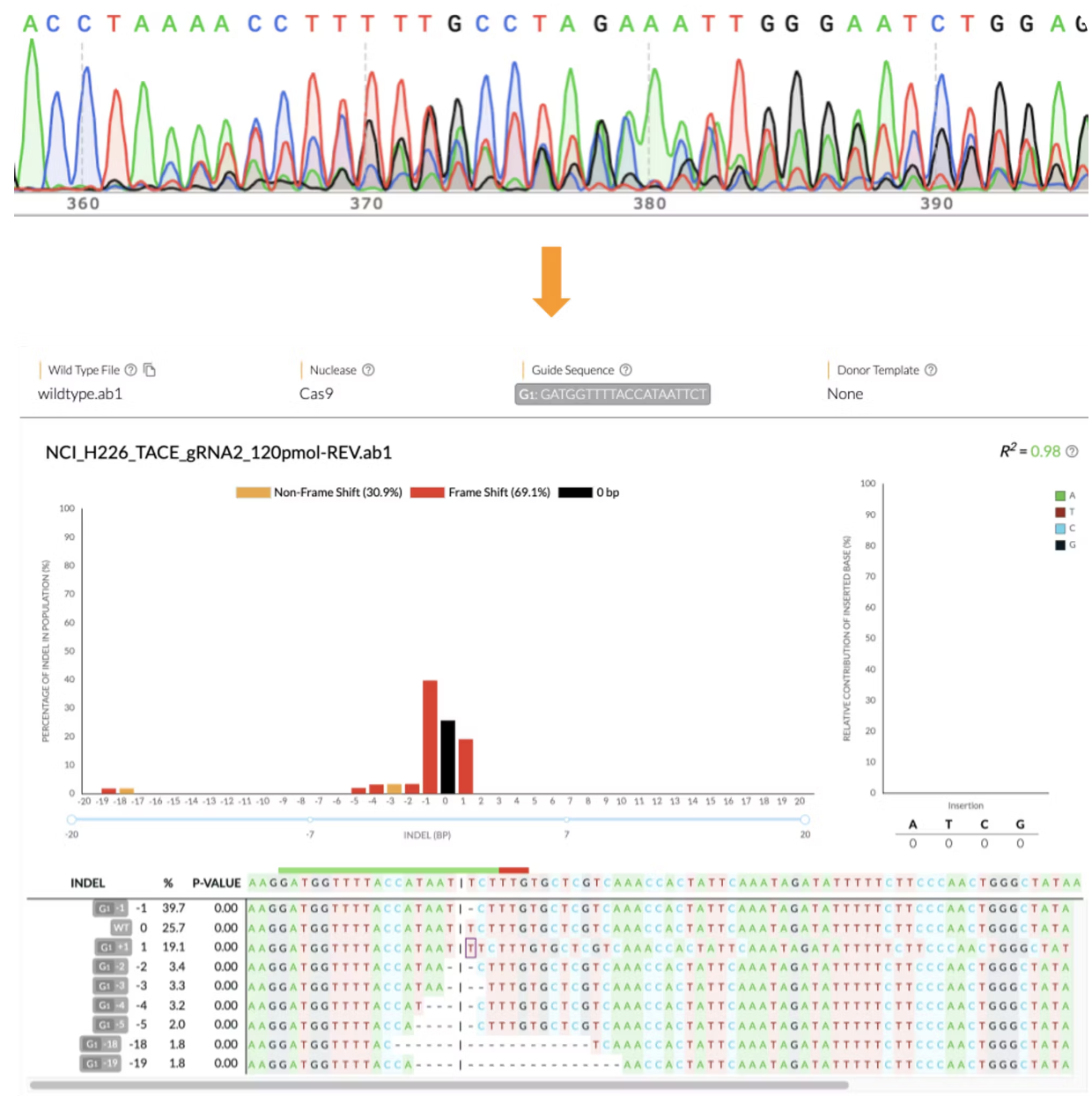
Raw Outputs
- donor_alignment.txt
- donor_insert.ab1
- logs.txt
- results.xlsx
- single_t_insertion.ab1 a. logs.txt b. results.xlsx
- dropout.ab1 a. logs.txt b. results.xlsx
- high_edit.ab1 a. logs.txt b. results.xlsx